This is why it is impossible to give a general protocol for the purification of proteins. But I can give a few guidelines:
- If nothing is known about the protein start with a buffer with neutral pH. Add 50 mM NaCl to increase the solubility of the protein. If possible avoid phosphate buffers as phosphate will give salt crystals in the crystallization screen. Phosphates can bind to nucleotide binding sides and are co-purified.
- Recombinant proteins with affinity tags often improve the yield and the purity. Because the purification is based on the properties of the tag and not the protein, check that the protein is correctly folded (Size-exclusion chromatography). If the affinity tag has to be removed before crystallization needs to be tested.
- Size-exclusion chromatography should be used as the last step in the purification. Check that the protein peak is at the right molecular weight, and that the profile is symmetric, without shoulder or tailing. Collect the fractions and check if possible for enzyme activity. Also measure the optical dencity 280/260 ratio, to check if the protein is contaminated with nucleotides.
- Enzymatic or functional assay.
- Purity of the protein sample.
- Sample homogeneity.
- Protein concentration and starting buffer.
Never mix batches from different purifications, due to small changes in the purity the optimal crystallization condition for each batch of protein can be different. If you don't purify the protein, ask for a copy of the purification protocol for every batch. Each batch optimally contains more than 10 mg of protein. If the protein can be stored safely at -20°C or -80°C, store the protein in aliquots to prevent multiple freeze-thaw cycles.
Purity of the protein sample.
(SDS page and IEF)
Different amounts of protein sample are loaded on the gel to check for contaminants and degradation products.
- Marker
- Large amount is loaded, contaminants are visible.
- Normal amount is loaded, protein looks pure.
- Low amount is loaded, degradation product is visible.
Crystals can occasionally be grown from impure samples, but size and quality can improve as the purity increases. If the protein contains contaminants and large amounts are available, further purification is recommended. But if the sample contains minor impurities and/or only a few milligrams of protein is available, try to find initial conditions for crystallization. Further purification can be used to optimize the crystals. Protein fragments by proteolysis can be a reason for failure or for crystals of a protein fragment. Change the protein purification (add protease inhibitor).
Sample homogeneity.
Avoid heterogeneity by optimizing the protein purification. If a purification protocol is changed, check the crystallization behavior by repeating the initial screen.
- Chemical heterogeneity (using mass spectrometry): proteolysis, post translational modifications.
(glycosylation, phosphorylation, methylation, oxidation).
- Add protease inhibitors during purification. Or check if an active and stable proteolytic fragment is formed, use this for crystallization.
- Add Dithiothreitol (1-10 mM DTT) or 2-mercaptoethanol.
- Change expression system.
- Separate phosphorylation products with IEC.
- Conformational heterogeneity (using dynamic light scattering): oligomerization, aggregation, denaturation.
- Change pH, temperature or add salt.
- Protein modification (site directed mutagenesis).
- Contamination of the sample with small molecules. Ions remaining from former purification steps can be responsible for irreproducibility in the crystallization results, or salt crystals.
Protein concentration and starting buffer.
- The recommended protein concentration is 5-20 mg/ml. The protein concentration is measured with a protein assay. If the protein is insoluble change the pH, ionic strength or salt.
- The buffer concentration in the protein solution should be 10-20 times lower then the buffer concentration used in the well (5-10 mM).
- The salt concentration should be as low as possible. Drops with PEG will increase in size when their is to much salt in the protein solution if high salt in the protein solution is needed. Add salt to the well after mixing the protein with the well solution. Check the solubility of the protein with different salts.
- Store the protein at 4°C after the dialysis against the start buffer, or in small aliquots in the freezer. Check protein stability after storage.
- Centrifuge or filter the protein solution before use to remove precipitated protein.
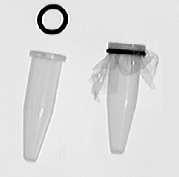
A piece of dialysis membrane (width 25 mm, length 50 mm) is soaked in water and cut to get a single layer of dialysis membrane (50 * 50 mm).
Pipet the sample in the Eppendorf tube (without lid) or other small tube.
Cover the tube with the dialysis membrane and place the O-ring (O-ring from dialysis button).
With a piece of parafilm the O-ring and membrane can be fixed.
Turn the tube upside down and check that the protein sample is in contact with the dialysis membrane.
With a piece of tape the dialysis set-up is placed in the buffer.
© 20-08-2018 Johan Zeelen